Neuroimaging Tutorials
The goal of this section is to allow basic exploration of resting state functional MRI data. Hopefully you will be able to get an idea of the range of complex and fascinating results that can be seen with some well considered data analyses. I've emphasised simple command line code, using freely available software, taking advantage of pre-compiled pipelines that can be run on a regular laptop. Much of the starter code is below, or everything is on my github page ready to download.
Finally, here are two papers that serve as a good introduction to some of the methods described below with examples of the expected results one can expect:
Advanced Normalisation Tools (ANTs)
I've just finished explorting Advanced Normalization Tools (ANTs) for registration and other pre-processing steps. In my opinion this is a huge leap forward and pretty much obfuscates many of the tricky issues in dealing with scans that have pathology in them e.g. tumours. However, it is slightly more complicated to use (e.g. it requires compiling from binaries) but hopefully the functions I've written will help a little. In summary: stick with the basic code on this site to start with, but feel free to follow this up with ANTs to get an idea of current state of the art methods or if you are looking to publish
A selection of functions to use ANTs on scans with tumours is on my github page.
NB: this code is to be enjoyed. All comments are welcome, but please see the individual licenses, and note that unfortunately I don't have the resources to offer follow-up support (sorry).
pre-processing
Standard resting state functional MRI requires some pre-statistical processing prior to analysis. How one does this can involve many separate functions and become rather complex: fortunately, there are now many excellent pipelines available that make this process far more enjoyable and robust. In addition, I've included some tips to help make masks as well as improving brain extraction and registration. The steps can be roughly broken down into:
1. Functional MRI pre-processing: This is mainly to reduce artifacts, improve signal quality, and help validate statistical assumptions.
2. Making masks: to define the tumour boundary
3. Brain extraction: this is helpful for registration, statistical analysis, viewing results, and other things
4. Registration: align images to each other to allow comparison between individuals
Here is a summary of the following stages:
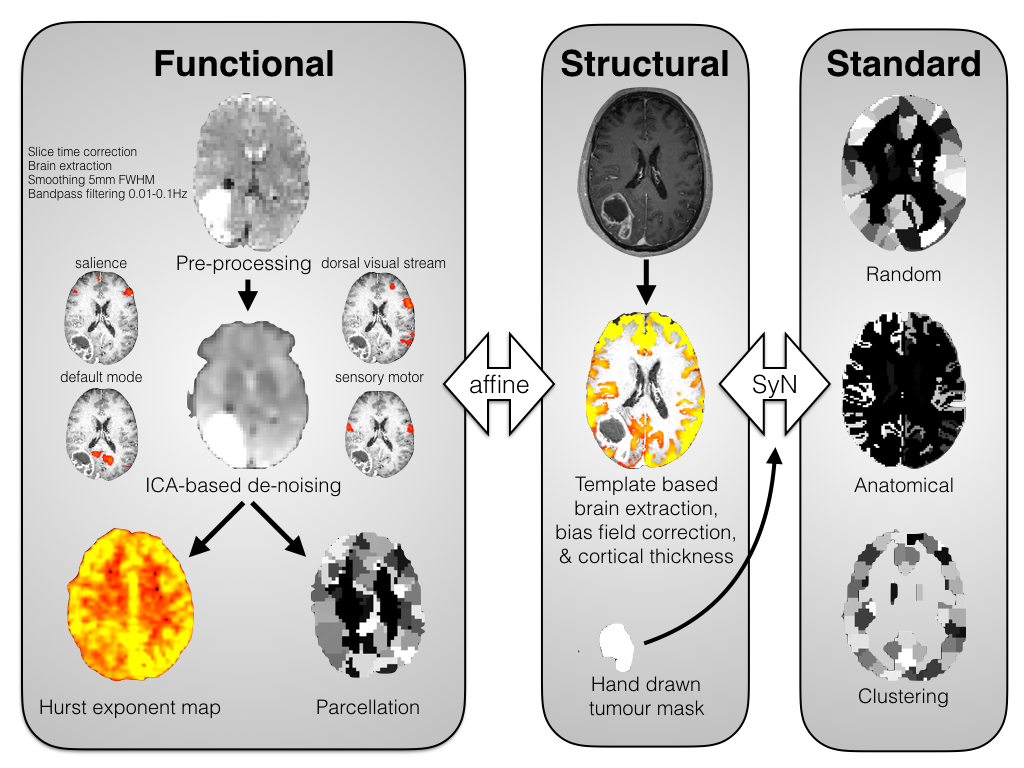
# resting FMRI pipelines
For single echo functional MRI I would recommend:
The website and toolbox has an helpful user guide which explains everything in an intuitive and thorough manner, including how to install the various required software packages (AFNI, FSL, MATLAB, Python). If you wish to read an example paper;
To use this package, an example command could be:
python ~/fmri_spt/speedypp.py -d coreg_tsoc.nii.gz -a mprage_brain_do.nii.gz --despike --tpattern=seq-z --zeropad=80 --rall --highpass=0.001
Selected options include:
- d
- a
--despike
--tpattern
--rall
--highpass
the single echo epi / resting functional MRI scan
the structural image / mprage
a novel approach for wavelet based de-noising
slice time acquisition
regress out....
frequency of the high pass filter
In Cambridge we have been using multi-echo resting state functional MRI coupled to independent component analysis. Again this requires a few dependencies to be set up but it also has a helpful user guide:
For single echo functional MRI I would recommend:
This is a novel technique that integrates image acquisition with subsequent de-noising in a physiologically principled manner. If you want to read more about the method and its development I would recommend checking out the following paper:
For single echo functional MRI I would recommend:
To perform ME-ICA, an example command could be:
meica.py -d "epi[1,2,3].nii.gz" -e 13,30.55,48.1 -b 15s --tpattern=seq-z --prefix=epi
Selected options include:
- d
- e
- b
- tpattern
- prefix
string of echo files to be concatenated e.g. epi1, epi2, …
corresponding echo times of the files above
removal of volumes/times at the start
slice time acquisition
string for flagging output files
Both of these pipelines have numerous other options: check out the documentation to make full use of their functionality. For example, other options include structural pre-processing (brain extraction and intensity normalisation) and registration: one could use these integrated options or use the following variations.
# masks
A mask is basically an image that selects out part of another image, just like a mask can cover up part of ones face. Here we specifically refer to a new image the outlines a specific lesion. This can be used to prohibit this area from being used in brain extraction and registation. If you are wondering why this would be useful, think about how a registration algorithm would try and match up a brain with a tumour to a healthy participant: we will also see that registration can be rather helpful for brain extraction too. Masks are also used for segmenting tissues and many other useful things, so they are an important concept to grasp.
To make a mask in fsl, do:
1. Load the image in the designated space that you want to make the mask into fslview
2. Select from the menu: File -> Create Mask
3. On the mode toolbar, click on the mask tool then the pen tool
4. Draw round the lesion on each image: whether you include the contrast enhancing part, oedema, or all areas affected by brain shift depends on what you are going to do with the image
5. On the toolbar, click on the fill tool and fill inside the border you have just drawn
5. Save the image: you will now have a mask of 1s inside the area you have drawn, and zeros elsewhere
# brain extraction
Standard automated brain extraction algorithims can struggle with images that are structurally abnormal. For example, the abnormal contrast around a tumour can cause difficulties in determining the boundary between grey matter and the surrounding CSF. Fortunately, a slightly different approach that uses a brain mask derived from groups of healthy participants can be used like a stencil to 'core out' the brain in a manner that is less sensitive to structural lesions: often this approach is sufficient to get a good brain extraction quickly and automatically. This approach has been described in detail by the Monti lab and they even have their own scripts available to download and a related publication:
I use a slight variation that uses linear rather than non-linear registration and a tumour mask: this appears to work better for tumours. The commands are:
1. Do a rough brain extraction:
bet mprage.nii.gz mprage_bet.nii.gz -B -f 0.1
2. Use this rough brain extraction to generate a transformation matrix to MNI space:
flirt -ref ${FSLDIR}/data/standard/MNI152_T1_2mm_brain_mask.nii.gz -in mprage_bet -inweight man_mask -omat bet2mni.mat
3. Invert the transformation matrix from step two to get a matrix to move from MNI space back to standard space
convert_xfm -omat mni2bet.mat -inverse bet2mni.mat
4. Use this inverse matrix to move an MNI space brain mask back to native/structural space
flirt -ref mprage_bet.nii.gz -in ${FSLDIR}/data/standard/MNI152_T1_2mm_brain_mask.nii.gz -init mni2bet.mat -applyxfm -out native_MNI_mask
5. Binarise this mask
fslmaths native_MNI_mask.nii.gz -bin native_MNI_mask.nii.gz
6. Use this binary mask to core out
fslmaths mprage.nii.gz -mas native_MNI_mask.nii.gz mprage_brain.nii.gz
7. Finally perform a final top up round of bet on this cored out brain
bet mprage_brain.nii.gz mprage_brain.nii.gz -f 0.1
It's really important to check the results from this step: this is a good check command to run:
slices mprage.nii.gz -s 2 mprage_brain.nii.gz
# registration
Registration lines up two images. At the very least the functional and structural images should be aligned: additionally, one could also register the images to a ‘standard’ space (such as MNI space). This latter step has the benefit easily allowing comparison of spacial networks (such as from SCA or ICA) by aligning them with templates that are also in standard space. However, for connectome analysis registration to a standard space isn’t always necessary as one could parcellate in ‘native space’ (the space that the images were acquired in) then use pre-defined standard space co-ordinates of the parcellation template.
Registration itself can be broken into four main steps:
1. Define spaces (structural = space of structural image taken directly from scanner; functional = space of functional image taken directly from scanner; standard = canonical registration space, based on for example the average of scanning multiple participants). One may also here the term 'native space' which refers to the space where the scanner acquired the images (and can therefore be structural or functional),
2. Choose a model to perform the registration (loosely based on the degrees of freedom the input is allowed to move to align to the reference image)
3. Specify a cost function to evaluate how well the images have been aligned
4. Apply an interpolation method to ‘fill in the gaps’ when moving images between spaces
For registration, I prefer FSL tools and keeping things as simple as possible. For brain tumours, often using simpler registration tools is better. For example, the presence of a tumour often means the priors specified for non-linear registration are not applicable, and boundary based registration is limited by loss of contrast changes due to the tumour. Using an affine linear registration (e.g. with FLIRT in FSL) with a simple binary mask for the tumour usually gives more than adequate results. To perform registration of both structural and functional scans to MNI space, do:
1. Create an affine registration of the brain extracted structural image to MNI space
flirt -ref ${FSLDIR}/data/standard/MNI152_T1_2mm_brain.nii.gz -in mprage_brain -omat struct2standard.mat \
-out mprage_MNI -inweight masks -interp nearestneighbour
2. Create a transformation matrix for the functional image to the original structural image in anatomical space
flirt -ref mprage_brain -in epi -dof 6 -omat func2struct.mat
3. Concatenate both these transforms from the last two steps to into one (careful with order here)
convert_xfm -omat func2standard -concat struct2standard.mat func2struct.mat
4. Apply the concatenated transform to all the 3D volumes of the functional image
flirt -ref ${FSLDIR}/data/standard/MNI152_T1_2mm_brain.nii.gz -in epi -applyxfm -init func2standard.mat -out epi_MNI
5. It’s really important to do a manual inspection of the quality of this step – a bad registration could nullify any subsequent analysis, and even with these options that are designed to help it work with abnormal structural images, it may still fail depending on the original data. Using a red outline of one image on another is a really good way.
slices mprage_MNI -s 2 epi_MNI
You should now have pre-processed resting state FMRI data registered to a brain extracted structural image both in MNI space. The main options selected here are:-ref
-in
-omat
-out
-inweight
-interp
-dof
-applyxfm
-init
the reference image (and corresponding space) to which we wish to match our input image
the input image we are matching to the reference image
the transformation matrix to go between image spaces
output image name (nb: only a single 3D volume)
tumour mask created earlier
interpolation method: nearest neighbour is *probably* recommended with lesions
degrees of freedom: for functional to structural often less is more
uses FLIRT to apply a transformation matrix i.e. move an image between spaces
the matrix used to move between spaces
seed connectivity analysis (SCA)
This depends on having AFNI & SUMA successfully installed - please see the respective websites for how to do this and for any support:
Here is a summary of SCA methods:
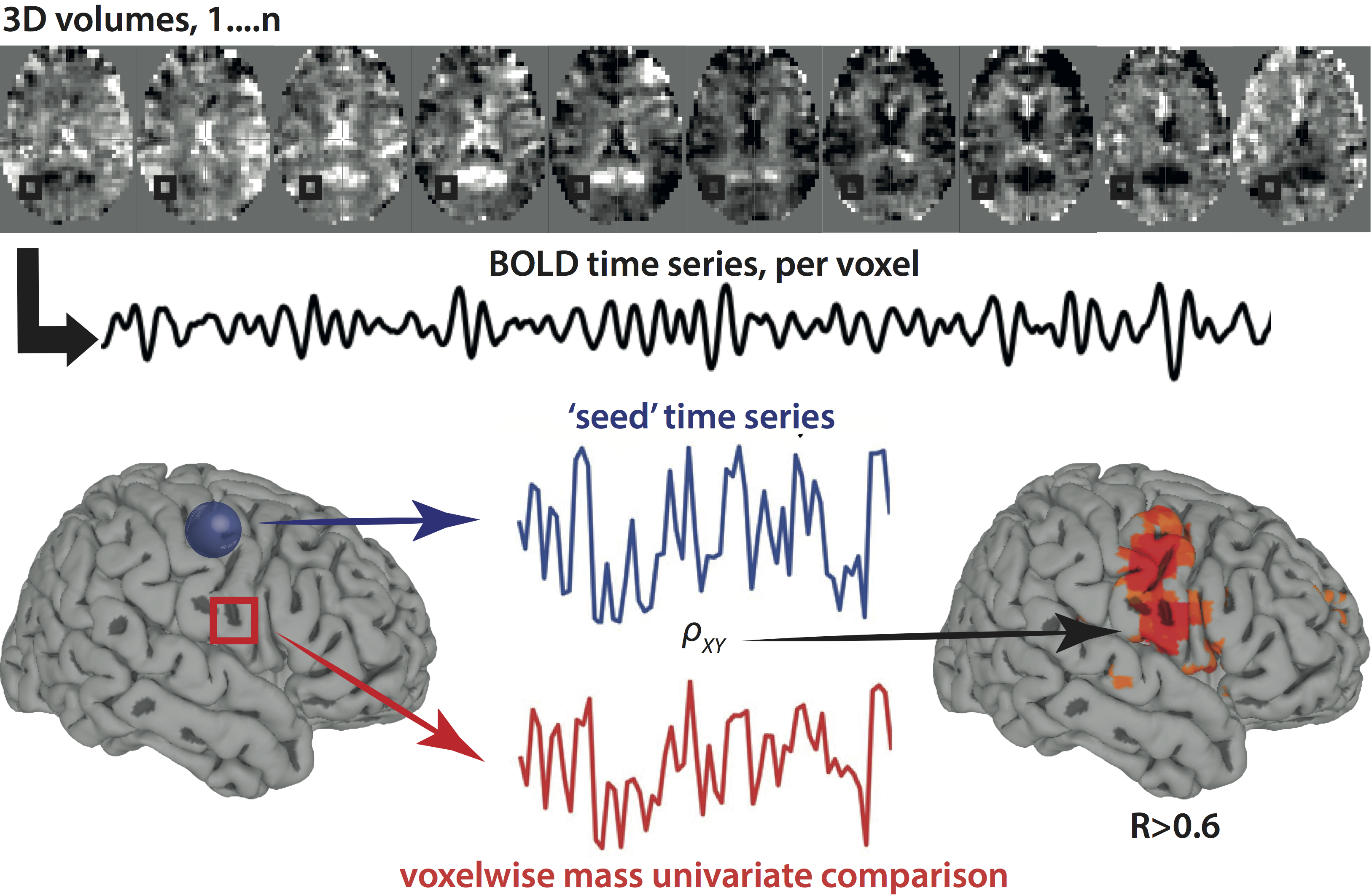
And here is a summary of some canonical resting state networks one can detect with this method:

Here is a paper describing these methods in more detail and some of the expected results:
To get started, you need:
1. EPI file in MNI space (see pre-processing tutorials)
2. MNI template (already in AFNI)
3. specific .spec files for mapping EPI to surface (in SUMA folder already)
4. add the EPI & MNI templates to the SUMA folder and cd into this
Then the steps are:
1. Convert your .nii.gz image to BRIK/HEAD files for AFNI
3dcopy originalfile.nii.gz -prefix newfile
2. Change to Talairach space for viewing
3drefit –view tlrc –space MNI newfile(i.e. output from step 1)
3. Load AFNI (with neuroimaging markup language to link with SUMA)
afni –niml &
4. Set files in AFNI
underlay = MNI template (e.g. MNI_N27 in SUMA folder)
overlay = newfile (output of 3dcopy in step 1 above)
5. Load SUMA (spec = hemisphere files, sv = surface volume
suma –spec N27_lh.spec –sv MNI_N27+tlrc.
6. Press 't' for talk, then '.' or ',' to switch between surfaces
7. Run InstaCorr in AFNI. You will need to select the output file from step 2 as your dataset. You can adjust correlation threshold and other parameters as you like: I would suggest a 5mm blur and keep all the others as default to start. Then, just move the mouse around to select the seed region and highlight the surrounding network. For more on running InstaCorr, see here:
And this is what is looks like when you put it all together. See the cursor moving across the cortex on the left screen. On the right screen is a pial inflated cerebral hemisphere (note this is a normative brain, so no tumour). On this screen the highlighted areas are the functional connectivity networks corresponding to the seed, which is the cursor location on the left screen. Note how the network fluctuates depending on the cursor location, and how at times it displays a variable likeness to canonical networks.
independent component analysis
Independent component analysis (ICA) is a pretty neat data driven approach to analysis. Doing this with Melodic (by FSL) is really nice as it is well principled, explained nicely (with great tutorials), and integrates well with viewing and de-noising. In fact, this would be my preferred way of analysing resting state functional MRI data in general is it produces great de-noised data for further analyses (including connectomics/graph theory), pulls out many of the canonical networks found with SCA, and also finds many more networks which can then be analysed as a hierarchical whole brain network (including integration with matlab and some nice visualisation using d3.js). NB: throughout I will refer to individual components (i.e. the output of ICA) as networks.
Here are some links to Melodic on the FSL site for further reference:
To get started the easiest way is to use the GUI (indeed this is the only way to include pre-processing at the same time). Options I would select (in addition to the defaults) are:
- Delete 2-3 volumes to allow steady state imaging to be achieved
- Slice time correction (unless you have a very short TR)
- No registration or resampling (we can do this with the code above or ANTs)
- Output full stats
If one already has pre-processed data through some other means then this can be run through melodic on the command line, bypassing any pre-processing (and here mainly sticking to default options):
melodic -i epi.nii.gz -o melodic --report -v
Finally, if one has a batch of scans one wishes to process in the same way, one can set up the GUI then save the output to run on the command line:
feat melodic_gui_save.fsf
Now (c.2-3 hours) we have our finished ICA data and a bunch of networks (c.80). We can create a nice summary of the data with the following command:
slices_summary melodic_IC_MNI 4 mprage_MNI melodic_IC.sum
Where melodic_IC is the 4D volume of all ICA networks created by melodic that has been registered to MNI space. NB: this is useful for some FSLNets stuff later so worth running.
We can check the concordance (spatial cross correlation) of our networks with canonical networks e.g. from healthy controls - this helps provide an objective way of classifying the networks:
fslcc -t 0.2 --noabs canonical_networks melodic_IC_MNI
Where canonical_networks represents the standards for network comparison - freely available templates are available here:
The next step will involve inspecting all the components manually to enable classification as either functional or noise related components. Classification within functional networks (i.e. those of neuronal origin) has been covered above: identifying noise components allows them to be removed from the data (known as "cleaning"). To classify noise components one needs to look at their spatial location and power spectra. Currently this is *probably* best done using Melview (a beta code programme downloadable from the FSL website) but in the next FSL update (version 6) due this summer I believe the new fslview (FSLeyes) should also be able to do this. In the meantime try the following command in fslview to visualise all the networks:
fslview mprage_MNI melodic_IC_MNI -b 5,15 -l "Red-Yellow" &
(the -b option allows appropriate thresholding for viewing, but feel free to experiment)
One we have labelled each individual component as good or bad (depending on spatial appearance and power spectrum, a 20 min task or so), we can remove them from our data to clean it up:
fsl_regfilt -i filtered_func_data -d melodic_mix -f XXX -o filtered_func_data_clean
Here XXX refers to the individual numbers of each component one wishes to remove (remember to check how they are counted in the files versus fslview). There are automatic ways of doing this but individual subjects with tumours are likely so heterogenous to either not be applicable for pre-trained component removal (cf. AROMA) or to allow training of a suitable sized cohort for machine learning approaches (cf. FIX). However, I'm happy to be proven wrong here (as always).
This data is now *perfect* for connectome/graph theory analysis (see below).
Finally, if you want to make some *sweeter* pictures, try upsampling into a higher resolution standard space:
<>for i in {1..nComponents}; do
flirt -in thresh_zstat${i}.nii.gz -ref MNI152_T1_0.5mm -applyxfm -usesqform thresh_zstat${i}_highres.nii.gz
done
graph theory
Once the imaging data has been processed effectively (see above) it's time to get down to connectomics and graph theory. One can do this in any number of software packages: I've chosen R in this instance as it's free, has a good interface to get started with in the form of RStudio, and includes some excellent libraries such as BrainWaver. More recently I've started using Matlab and will be releasing some code for that too. The following R code is designed to allow one to replicate the figures from my recent paper:
The actual files are all available via my github page: just make a copy, set the paths to the data, and run.
And here is a brief summary of the methods:
